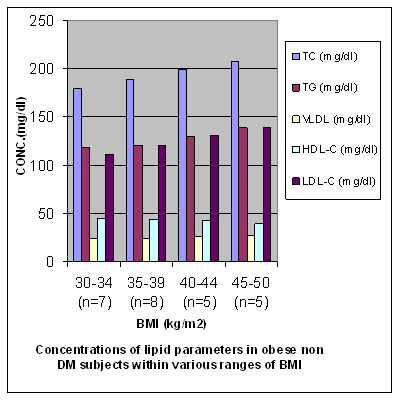Electronic Journal of Integrative Biosciences
|
Electronic Journal of Integrative Biosciences |
|
a and b, a* and b* show P < 0.001, a and c, a* and c* show P < 0.0001
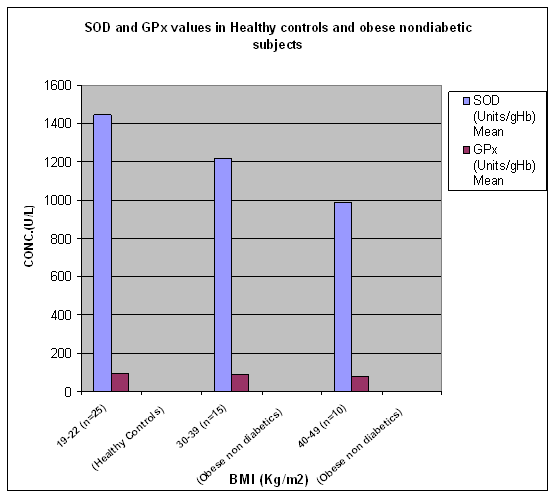
Figure 1
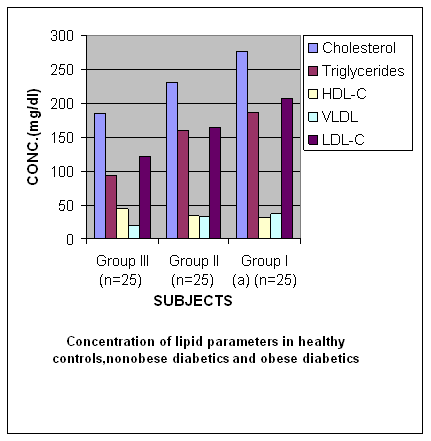
Table 2Levels of various Biochemical Parameters assessed in Lipid Profile in Various Groups of Subjects (Healthy Controls, Non Obese type 2 Diabetics and Obese type 2 Diabetics)
º vs* P < 0.001(Very significant) * vs #, º vs # P < 0.0001(Highly significant) Figure 2
Table 3Concentrations (expressed as mean ± SD) of lipid profile parameters in obese non diabetic subjects (group IIb) in various ranges of BMI
P values a Vs b TC, TG, VLDL, HDL-C, LDL-C are 0.04 (S), 0.55 (NS), 0.54 (NS), 0.30 (NS), 0.05(S) P values a Vs c TC, TG, VLDL, HDL-C, LDL-C are 0.004 (VS), 0.05 (S), 0.05 (S), 0.01 (S), 0.005 (S) P values a Vs d TC, TG, VLDL HDL-C, LDL-C are 0.0002 (HS), 0.002(VS), 0.002 (VS), < 0.0001 (HS), 0.0002 (HS). P values b Vs c TC, TG, VLDL, HDL –C, LDL-C are 0.04 (S), 0.05 (S), 0.05 (S), 0.03 (S), 0.05 (S). P values b Vs d TC, TG, VLDL, HDL-C, LDL-C are 0.0004 (HS), 0.0008 (HS), 0.0008 (HS), 0.0001 (HS), 0.0004 (HS). P values c Vs d TC, TG, VLDL, HDL-C, LDL-C are 0.03 (S), 0.03 (S), 0.03 (S), 0.006 (VS), 0.03 (S). S = Significant NS = Non significant VS = Very significant HS = Highly significant Figure 3
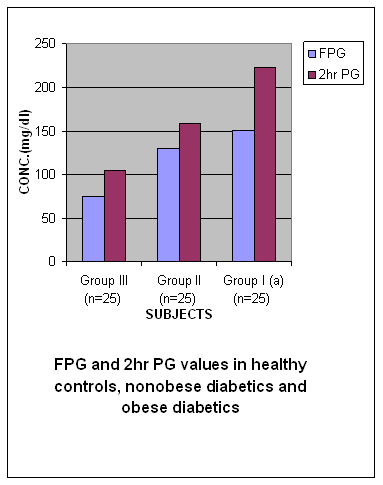
Table 4 Values of Fasting and Post Prandial Plasma Glucose (FPG, 2hr PG) in Healthy Controls, Non-Obese type 2 Diabetics and Obese type 2 Diabetics
* and # P < 0.0001 (Highly significant)
Figure 4
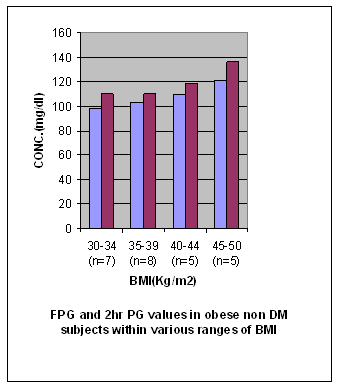
Table 5 Concentrations (expressed as mean ± SD) of FPG and 2hr PG in obese non diabetic subjects (group IIb) in various ranges of BMI
S = Significant NS = Non significant VS = Very significant HS = Highly significant Figure 5
Results The results of the study that is values of antioxidant enzymes SOD and GPx, Lipid profile and Glucose in the subjects are given in tables 1-5 respectively and in figures1-5. Table 1 (Figure1) shows an inverse relationship between BMI and erythrocyte SOD activity. The mean erythrocyte SOD activity of subjects with healthy body weight (1443.45 ± 176.84 units/g Hb) was significantly higher (P<0.001) than in subjects with BMI greater than 30 kg/m2 (1218.0 ± 31.0 units/g Hb). The table also shows that subjects with BMI above 40 kg/m2 have the lowest activity of erythrocyte SOD (986.0 ± 25.0 units/g Hb). Table 1 also shows an inverse relationship between the erythrocyte cytoprotective enzyme GPX and BMI. The activity of this enzyme in individuals with healthy BMI (95.1±3.6 units/g Hb) was significantly higher (P<0.001) than the value in those with BMI greater than 30 kg/m2 (92.3±3.2 units/g Hb). Subjects with BMI greater than 40 kg/m2 had the least activity of this enzyme (80.8 ± 7.2units/g Hb). Thus the results support the role of obesity in decreasing the activities of antioxidant enzymes or in other terms enhancing oxidative stress. In Table 2 (Figure 2) the values of various biochemical parameters measured in lipid profile showed a highly significant pattern in group II (p< 0.001) and Group I(a) (p<0.0001) subjects compared to healthy controls. Also, the values were significantly higher (P < 0.0001) in group I (a) compared to group II supporting the effect of obesity in further impairing the lipid levels in type 2 diabetics. In Table 3 (Figure 3) the values of TC, TG, VLDL, HDL-C and LDL-C showed significantly increasing patterns with increase in BMI. Significantly high values for TC and LDL-C were obtained in group IIb subjects with BMI 35-39 Kg/ m2 compared to those with BMI 30-34Kg/ m2 (0.04, 0.05).Further, subjects with BMI 40-44 Kg/ m2 showed significantly high values for all lipid parameters (0.004, 0.05, 0.05, 0.01, 0.005) compared to those with BMI 30-34Kg/ m2.Significant differences in values were obtained in subjects with BMI 45-50 Kg/ m2 (0.0002, 0.002, 0.002, < 0.0001, 0.0002) compared to those with BMI 30-34Kg/ m2. Even when subjects with BMI 35-39Kg/ m2 were compared to those with BMI 40 - 44 Kg/ m2 significant differences in values were obtained (0.04, 0.05, 0.05, 0.03, 0.05). Further, on comparing subjects with BMI 35-39Kg/ m2 with BMI 45 - 50 Kg/ m2 significant differences in values were obtained (0.0004, 0.0008, 0.0008, 0.0001, 0.0004).Values for subjects with BMI 45 - 50 Kg/ m2 were significantly higher when compared to subjects with BMI 40-44 Kg/ m2 (0.03, 0.03, 0.03, 0.006,0.03). In table 4 (figure 4) the plasma glucose values (FPG and 2hr PG) were significantly higher (P < 0.0001) in group I (a) compared to group II reflecting the effect of obesity in impairing the glucose levels further, in subjects with type 2 diabetes. In table 5 (figure 5) the FPG and 2hr PG values in obese non diabetic subjects with BMI 40-44 Kg/ m2,45-50 Kg/m2 showed significantly higher values compared to subjects with BMI 30-34 Kg/ m2 [FPG:0.03, 0.001 ;2hrPG: 0.08(not quite significant),0.0005]. Further, 2hr PG values showed a significant pattern in subjects with BMI 40-44 Kg/ m2 compared to those with BMI 35-39 Kg/ m2(0.05) and 45-50 Kg/ m2(0.03).FPG and 2hr PG values were higher in subjects with BMI 45-50 Kg/ m2 compared to subjects with BMI 35-39 Kg/ m2 (0.002, 0.0002). Discussion The obesity epidemic is of considerable importance since it runs parallel to the type 2 DM and metabolic syndrome epidemic, we are currently experiencing. It is important to single out obesity as it plays an important role in the development of abnormalities related to glucose and lipid metabolism22. In our study we noted a decrease in levels of antioxidant enzymes viz. SOD and Gpx with increase in BMI. The decrease was significant in subjects with BMI 40-49 Kg/m2 (P < 0.0001) compared to subjects with BMI 19-22 Kg/m2. The decrease in SOD and Gpx activities in obese could be due to increased H2O2 production in adipose tissue of obese. SOD is believed to play a major role in the metabolism of reactive oxygen species (ROS). It is the first enzyme involved in the destruction of superoxide (O2-) anion radicals. It converts O2- into hydrogen peroxide (H2O2). Animal cells contain two intracellular forms of SOD, the cytoplasmic or copper zinc form (Cu – Zn SOD) and mitochondrial or manganese form (Mn-SOD). This enzyme is the first line of defense against O2- anion radicals and can be induced rapidly in some conditions such as exposure to oxidative stress (OS) of cells or organs23, 24. H2O2 is metabolized by Gpx in synergy with glutathione reductase (GSH). Gpx has a much higher affinity for H2O2 than catalase suggesting that H2O2 is mainly degraded by Gpx under normal condition23,24.Furukawa et al., (2004) suggested that adipose tissue is the major source of elevated plasma ROS. In normal conditions a state of redox homeostasis is present which is the normal physiologic process of reduction and oxidation in order to repair unstable, damaging ROS which include toxic oxygen free radicals [O2- and OH- (hydroxyl radical)], the highly unstable pro-oxidant oxygen non radicals (H2O2, singlet oxygen and organic analogues). OS implies a loss of redox homeostasis with an excess of ROS. OS is associated with an overproduction of ROS as well as an impairment of antioxidant defensive capacity as found in type 2 DM, metabolic syndrome and obesity alone.25 Obesity increases the OS by three possible mechanisms. Firstly, it increases the mechanical and metabolic load on the myocardium, thus increasing myocardial oxygen consumption. A negative consequence of the elevated myocardial oxygen consumption is the production of ROS such as O2-, hydroxyl radical and hydrogen peroxides from the increased mitochondrial respiration. Leakage of electrons out of the mitochondrial election transport chain promotes a one electron reduction of molecular oxygen resulting in the formation of O2- radicals.26 The second mechanism by which obesity can independently cause OS is by progressive and cumulative cell injury resulting from pressure from the large body mass. Cell injury causes the release of cytokines especially tumour necrosis factor alpha (TNF- α) which generates ROS from the tissues 27. A third possible mechanism is through diet which is probably a predominant cause in India. Nutritional obesity implies the consumption of hyperlipidemic diets which may be involved in oxygen metabolism. Double bonds in the fatty acid molecules are vulnerable to oxidation reactions and may consequently cause lipid peroxidation.28 Thus, the decreased values of SOD and GPx observed in our study could be due to increased OS caused by obesity which causes stimulation of antioxidant enzymes. But over a period of time the stores of antioxidant enzymes are depleted and cannot keep pace with increasing OS. In our study we also observed a significant increase in TC, TG, VLDL-C, LDL-C values (P = 0.0002, P = 0.002, P = 0.002, P = 0.0002) in obese non diabetics with BMI 45-49 kg/m2 compared to those with BMI 30-34 kg/m2. Further, comparison of obese diabetic subjects (Group IIa) with obese non diabetics (Group IIb) yielded significant results suggesting that with obesity insulin resistance worsen and dyslipidemia in type 2 diabetics impairs further29, 30. This causes raised TC, TG, VLDL-C, LDL-C and lower levels of HDL-C in obese diabetics compared to obese non diabetes. According to Grundy (2005) dyslipidemia associated with obesity is multi-factorial, and is frequently associated with a cluster of interrelated cardiovascular disease risk factors. Obesity is a critical determinant of dyslipidemia and operates through a number of metabolic influences that include reduced insulin sensitivity and changes in fatty acid metabolism. Variations in the nature and magnitude of the dyslipidemia are due to the interaction of genetic factors with environmental influences most notably diet and physical activity, and possibly stress31. Among the major effects of excess adiposity on plasma lipoproteins are increases in levels of TG rich VLDL particles. Both adipocyte derived fatty acids and cytokines, or adipokines, can promote increased TG synthesis, leading to increased hepatic secretion of TG enriched VLDL. Plasma VLDL levels can increase further as a result of reduced lipolysis and clearance due to the lower peripheral activity of lipoprotein lipase (LPL) associated with adiposity. Partially lipolysed VLDL remnants can then return to the liver, adding to the TG pool available for VLDL secretion32. As TG level increases in obese individuals the diameter of major LDL species decline. The mechanism for increased LDL according to Berneis (2002) is that a higher level of plasma TG is associated with larger VLDL particles are lipolysed less efficiently by LPL. This gives rise to remnant particles. These remnants have increased content of the apoprotein C III. Their slow lipolysis lead to reduced receptor mediated plasma clearance33. The remnants are further lipolysed by the combined action of LPL and hepatic lipase (HL) a process mediated by cholesterol ester transfer protein (CETP). The resulting TG is, in addition delipidated and remodeled to form smaller, lipid depleted LDL. These particles have lower affinity for LDL receptor. Moreover, higher levels of remnant particles lead to increased exchange of TG for cholesterol in both LDL and HDL, a process mediated by CETP. TG rich LDLS and HDLS are degraded further by HL, leading to yet smaller LDLs and to smaller and less stable HDLs that are more rapidly catabolised, resulting in reduced HDL cholesterol (Fig. 6). There is a strong negative correlation between obesity and HDL-C levels with a decrease of approximately 0.4 mg/dl of HDL-C with each kg/m2 increment of BMI33, 34. Factors causing dyslipidemia: · High carbohydrate diet · Adiposity · Insulin resistance · Genetic predisposition Fig 6: DYSLIPIDEMIA OF OBESITY
Liver Larger VLDL Remnants Small LDL
TG
Smaller HDL Smaller LDL
CETP = Cholesteryl ester transfer protein HDL = High density lipoprotein LDL = Low density lipoprotein HL = Hepatic lipase Chol = Cholesterol TG = Triglycerides LPL = Lipoprotein lipase
According to Williams (1997) increased BMI is associated with shift from larger to smaller LDL particles. They suggested an association of body fat with atherogenic dyslipidemia35. This is consistent with the findings of our study. We also observed an abnormal lipid profile with increasing BMI values in obese non diabetics. In a recent study by Redinger (2007) excessive storage of fatty acid as TG within adipocytes creates obesity. This eventually leads to the release of excessive fatty acids from enhanced lipolysis, which is stimulated by the enhanced sympathetic state existing in obesity36. The release of these excessive free fatty acids (FFAs) then incites lipotoxicity, as lipids and their metabolites create oxidant stress to the endoplasmic reticulum and mitochondria. This affects adipose as well as non adipose tissue, accounting for its pathophysiology in many organs such as the liver and pancreas, and in metabolic syndrome37, 38. The FFAs released from excessively stored TG deposits also inhibit lipogenesis, preventing adequate clearance of serum TG levels that contribute to hypertriglyceridemia39. The above mechanism probably causes insulin receptor dysfunction. This results in an insulin resistant state that creates hyperglycemia with compensated gluconeogenesis39, 40. The above explanation supports the results of our study as elevated FPG and 2 hr PG levels were observed in obese non diabetics. The obesity induced hyperglycemic state also increases hepatic glucose production. This accentuates the insulin resistance in obese as BMI increases. It is also decreases utilization of insulin stimulated muscle glucose contributing further to hyperglycemia39, 40. A significant pattern of increase in levels of FPG and 2 hr PG was observed in obese non diabetic subjects with increase in BMI from 30 to 50 kg/m2. This can be substantiated by the significant results obtained an comparing obese subjects with BMI 30-34 kg/m2 with subjects in BMI ranges 35-39 kg/m2, 40-44 kg/m2 and 45-50 kg/m2 respectively (P = 0.08, P = 0.03, P = 0.001 for FPG values) (P = 0.84, P = 0.08, P = 0.0005 for 2 hr PG)(Table5). The above results are in agreement with the following concept that resistance to insulin action augments with increasing severity of obesity. With passage of time the functional capacity of the insulin secreting cells of islets of langerhans first increases, as a result of hypertrophy and perhaps limited hyperplasia. When the total functional capacity of the system is reached the decompensation and perhaps true exhaustion of the insulin producing ability occurs. This results in a gradual yet continuous deterioration of glucose intolerance in the obese subjects who are likely to end up with type 2 DM41. The above mechanism illustrates the role of obesity in causing DM. However, also in our study the obese diabetic subjects (group IIa) showed a more atherogenic lipid profile and also significantly higher FPG and 2 hr PG values compared to obese non diabetic subjects. This suggests that the above discussed effects of obesity on lipids and glucose are amplified in diabetics with obesity. The decreased levels of antioxidant enzymes observed in obese non diabetics may also play a role in impairing glucose tolerance in these subjects. We have discussed previously that obesity causes increases in OS. It is known that OS impairs both insulin secretion by pancreatic β cells, glucose transport in muscle and adipose tissue 23. The possible mechanism could be that increased production of ROS is accompanied by augmented expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and decreased expression of antioxidative enzymes and OS. This causes dysregulated production of adipocytokines (fat derived hormones). Excessive and long term exposure to ROS reduces insulin sensitivity and impairs glucose as well as lipid metabolism23 (Fig. 7). Fig.7. ROS production in accumulated fat contributes to insulin resistance, DM and atherosclerosis
↑ NADPH oxidase ↓ Antioxidative enzymes
↑ ROS ↑ ROS
OS to remote tissues ↑ OS, dysregulation of adipocytokines Insulin resistance, Diabetes mellitus and Atherosclerosis Conclusion From the study it is indicated that obesity is an independent risk factor to cause decreased activity of antioxidant enzymes. Obesity is an exaggeration of normal adiposity and is a central player in the pathophysiology of DM, insulin resistance, dyslipidemia, hypertension and atherosclerosis. Obesity is a major contributor to the metabolic dysfunction involving lipid and glucose as well as in complicating the clinical symptoms in subjects already suffering from type 2 diabetes. Thus, it may be suggested that weight control is a prerequisite for an obese so as to avoid the associated metabolic complications. It is also of considerable concern in India as urbanisation has resulted in several changes in life style which is causing a clustering of cardiovascular risk factors namely central adiposity, obesity, hyperinsulinemia, dyslipidemia, hypertension and glucose intolerance.
Literature cited 1. U.S Dept. of Health and Human Service, National Institutes of Health “Clinical Guidelines on the Identification, Evaluation and Treatment of Overweight and Obesity in Adults”. The Endocrine Report (2000). NHLBI document 98 – 4083. 2. US Department of Health and Human Services. The Surgeons general report on Nutrition and Health US Department of Health and Human Services: Washington, DC, 1988, Department of Health and Human Services Publication 88-50210. 3 Manson JE, Stampfer MJ, Hennekens CH, Willet WC. Body weight and longevity. A reassessment. JAMA 1987; 257: 353-358. 4 Banerji MA, Faridi N, Atluri R et al. Body composition ,visceral fat, leptin and insulin resistance in Asian Indian Men. J clin Endocrinol and Metab 1999;84:1137 – 44 5 Ramachandran A, Snehalatha C, Kapur A, et al. Prevalence of diabetes and impaired glucose tolerance in India. National Urban Diabetes Survey. Diabetologia 2001; 44: 1094 – 1101. 6 Ramachandran A, Snehalatha C, Kapur A, Vinitha R et al. Prevalence of overweight in urban Indian adolescent school children . Diab Res Clin Pract.2002; 57:185 – 90. 7 Obesity. Preventing and managing the global epidemic, report of a WHO Consultation .WHO Technical Report Service 2000; 894:1 – 253. 8 Shimomura I and colleagues. Oxidants link obesity to diabetes .The Journal of Clinical Investigation, 2004; 114(12): 1752 – 1761. 9 American Association of Clinical Endocrinologists/ American College of Endocrinology (AACE/ACE). AACE/ ACE Position Statement on the Prevention, Diagnosis and Treatment of Obesity (1998 Revision). Endocrine Practice 1998; 4: 297-330. 10 National Institutes of Health Consensus Development Panel on the Health Implications of Obesity. Health implications of obesity: National Institutes of Health Consensus Development Conference Statement. Ann Intern Med 1985; 103:1073-7. 11 Ramachandran A. Epidemiology of diabetes in India – 3 decades of research. JAPI, 2005; 53: 34 – 38. 12 . Zimmet P. Kelly .Challenges in diabetes epidemiology from west to rest. Diabetes Care.1992; 15: 232 – 252. 13 Despres JP. Obesity and lipid metabolism: relevance of body fat distribution. Curr. Opin Lipidol 1991; 2: 5-15. 14 Albrink MJ, Krauss RM, lIndgren Ft and Wood PD. Intercorrelation among plasma High density lipoprotein, obesity and triglycerides in a normal population. Lipids, 1980; 15:668. 15 Caraway WT. Carbohydrates. In Fundamentals of Clinical Chemistry by Tietz N.W. ed , W.B. Saunders Co., Philadelphia PA, 1976: Chapter 6, page 243. 16 Meiattini F. et al. The 4–hydroxybenzoate / 4 aminophenazone Chromogenic system. Clin Chem.1978; 24(12): 2161 – 2165. 17 Buccolo G. et al. Quantitative determination of serum triglycerides by use of enzymes. Clin Chem.1973; 19(5):476 – 482. 18 Allain CC, Poon LS, Chan CSG, Tichmond W and Fu PC. Estimation of High density lipoprotein cholesterol by phosphotungstate Method. Clin Chem. 1974; 20:470 – 475. 19 Friedwald WT, Levy RI and Fredrickson DS. Estimation of concentration of LDL –C in plasma without the use of preparative ultra centrifuge. Clin Chem. 1972; 18: 499 – 502. 20. McCord JM, Fridovich I. Superoxide dismutase. An enzymatic function for erythrocuprein (hemocuprein). J Biol Chem 1969; 244: 6049-6055. 21. Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 1967; 70: 158-169. 22. Grundy SM: Metabolic complications of obesity. Endocrine. 13(2):155-165, 2000.
23. Fridovich I. Superoxide dismutase: regularities and irregularities. Harvey Lect 79: 51-75, 1985.
24. Viroonudomphol D, Pongpaew P, Tungtrongchitr R, Phonrat B, Supawan V, Vudhivai N, Schelp FP: Erythrocyte antioxidant enzymes and blood pressure in relation to overweight and obese Thai in Bangkok. South east Asian J. Trop Med. Public Health 31 (2): 325- 334, 2000.
25. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. The Journal of Clinical Investigation 114 (12): 1752-1761, 2004.
26. Turrens JF: Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17: 3-8, 1997.
27. Lechieitner M, Koch T, Harold M, Dzien A, Hoppiahler F: Tumour necrosis factor alpha plasma level in patients with type 1 diabetes mellitus and its association with glycemic control and cardiovascular risk factors. J Intern Med. 248: 67-76, 2000.
28. Moor de Burgos A, Wartanowics M, Ziemlanski S: Blood vitamin and lipid levels in overweight and obese women. Eur J Clin Nutr 46: 803- 808, 1992.
29. Alexander CM, Landsman PB, Teutsch SM: Diabetes mellitus, impaired fasting glucose, atherosclerotic risk factors and prevalence of coronary heart disease. Am J Cardiol 86; 897-902, 2000.
30. Lewis GF, Carpentier A, Adeli K, Giacca A: Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 23; 201-229, 2002.
31. Grundy SM: Obesity, metabolic syndrome and cardiovascular disease. J Clin Endocrinol Metab 89; 2595-2600, 2005.
32. Krauss RM, Siri PW: Metabolic abnormalities: triglyceride and low density lipoprotein. Endocrinol Metab Clin North Am 33: 405-415, 2004.
33. Berneis KK, Krauss RM: Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 43: 1363-1379, 2002.
34 Albrink MJ, Krauss RM, Lindgren FT, Wood PD. Intercorrelations among plasma high density lipoprotein, obesity and triglycerides in a normal population. Lipids 15:668, 1980.
35. Williams PT, Krauss RM: Associations of age, adiposity, menopause, and alcohol intake with low density lipoprotein subclasses. Arterioscler Thromb Vasc Biol 17: 1082-1090, 1997.
36. Redinger RN: Pathophysiology of obesity and its clinical manifestations. Gastroenterology and hepatology 3(11): 856-863, 2007.
37 Evans RM, Barish GD, Wang YX, PPARs and the complex journey to obesity. Nat Med 10; 355-361, 2004.
38 Hutley L, Prins JB.Fat as an endocrine organ: relationship to the metabolic syndrome. Am J Med Sci 330; 280-289, 2005.
39 Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA: Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46; 983-988, 1997.
40 Boden G, Chen X, Ruiz J, White JV, Rosseti L: Mechanism of fatty acid induced inhibition of glucose uptake. J Clin Invest 93: 2438-2446, 1994. 41 Rao GMM, Morghum LO.Relationship of obesity to diabetes. Horm. Metab.Res 16:209-210, 1984.
Accepted for publication 19 October 2008
|
|